


OUR MEDICINES
A range of medicines have been developed by Ferring in core therapy areas.
Which medicines have been developed?
Which medicines have been developed?


EUFLEXXA® is a viscoelastic, sterile solution of highly purified, high molecular weight (2.4-3.6 million daltons) hyaluronan (also known as sodium hyaluronate) in phosphate-buffered saline. EUFLEXXA® is indicated for the treatment of pain in osteoarthritis of the knee in patients who have failed to respond adequately to conservative non-pharmacologic therapy and simple analgesics (e.g. acetaminophen).
EUFLEXXA® was approved by the FDA in 2005.
Below is the key scientific publication that substantiated the FDA approval of EUFLEXXA® (sodium hyaluronate).
This product label was last updated on Jul - 2016
Each 1 mL of EUFLEXXA contains:
Sodium hyaluronate
Sodium chloride
Disodium hydrogen phosphate dodecahydrate
Sodium dihydrogen phosphate dihydrate
Water for injection
10 mg
8.5 mg
0.56 mg
0.05 mg
q.s.
EUFLEXXA is a viscoelastic, sterile solution of highly purified, high molecular weight (2.4-3.6 million daltons) hyaluronan (also known as sodium hyaluronate) in phosphate-buffered saline. EUFLEXXA is a very highly purified product extracted from bacterial cells. It is a polysaccharide consisting of repeating disaccharide of N-acetylglucosamine and sodium glucuronate, linked by alternating }⟶1,3 and }⟶1,4 glycosidic bonds.
Adverse event information regarding the use of EUFLEXXA as a treatment for pain in OA of the knee was available from two sources; a 12 week multicenter clinical trial conducted in Germany, and a 26 week multicenter clinical trial conducted in the US.
The most common adverse events related to EUFLEXXA injections reported in the clinical studies are the following:
All adverse events related to EUFLEXXA injections reported in Tables 1, 2, 3 and 4.
The following adverse events are among those that may occur in association with intra-articular injections
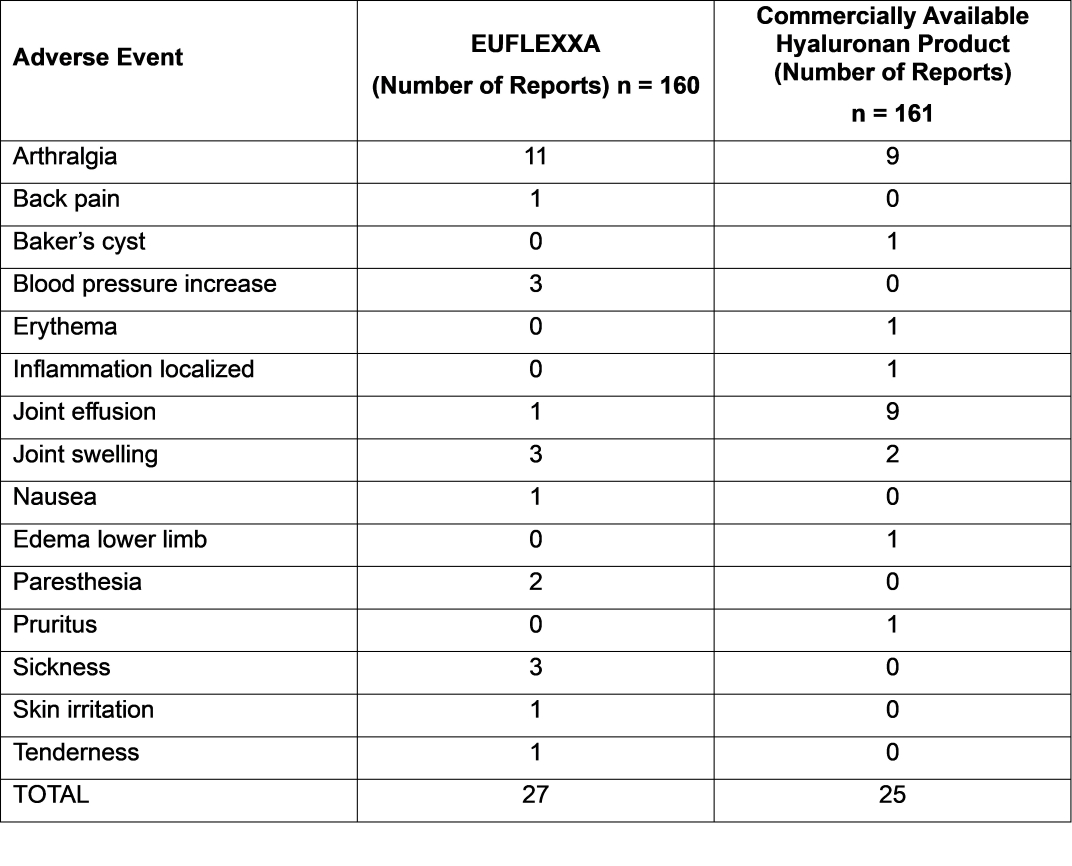
This clinical investigation was a prospective randomized, double-blinded, active control (commercially available hyaluronan product) study conducted at 10 centers. Three hundred twenty-one patients were randomized into groups of equal size to receive either EUFLEXXA (n=160) or the active control (n=161).
A total of 119 patients reported 196 adverse events; this number represents 54 (33.8%) of the EUFLEXXA group and 65 (44.4%) of the active control group. There were no deaths reported during the study. Incidences of each event were similar for both groups, except for knee joint effusion, which was reported by 9 patients in the active control group and one patient in the EUFLEXXA treatment group. Fifty-two adverse events were considered device-related. Table 1 lists the adverse events reported during this investigation.
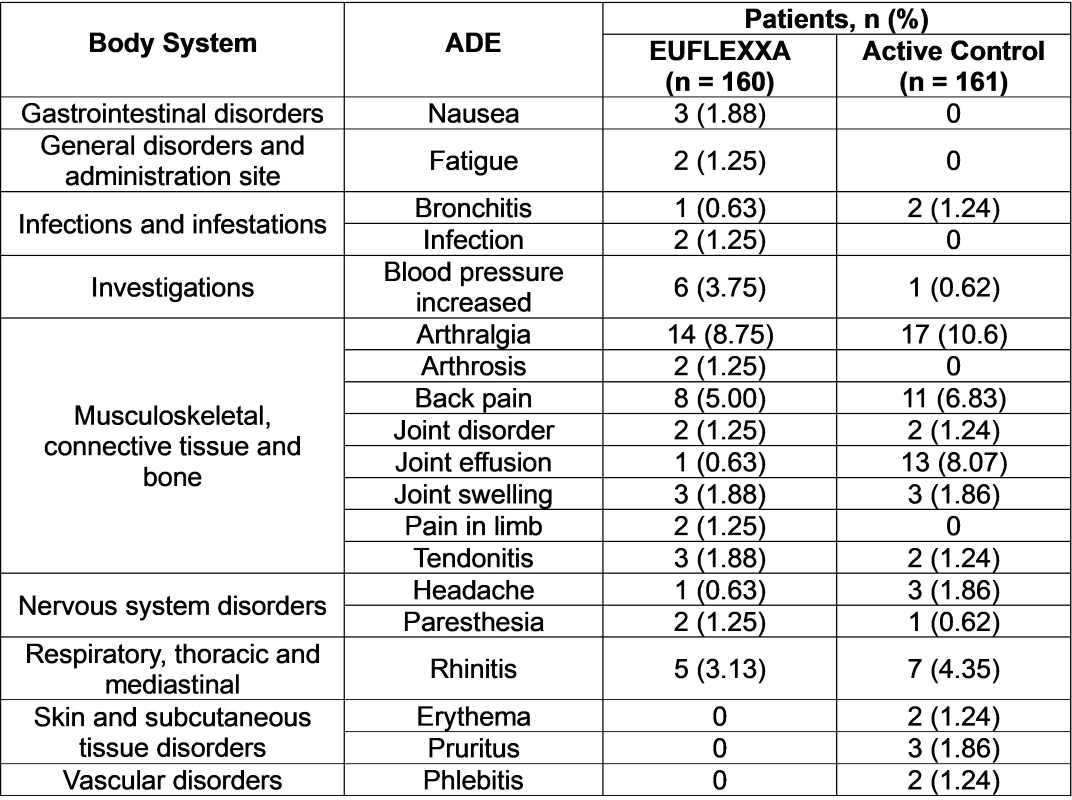
A total of 160 patients received 478 injections of EUFLEXXA. There were 27 reported adverse events considered to be related to EUFLEXXA injections: arthralgia – 11 (6.9%); back pain – 1 (0.63%); blood pressure increase – 3 (1.88%); joint effusion – 1 (0.63%); joint swelling – 3 (1.88%); nausea – 1 (0.63%); paresthesia – 2 (1.25%); feeling of sickness of injection – 3 (1.88%); skin irritation – 1 (0.63%); tenderness in study knee – 1 (0.63%). Four adverse events were reported for the EUFLEXXA group that the relationship to treatment was considered to be unknown: fatigue – 3 (1.88%); nausea – 1 (0.63%).
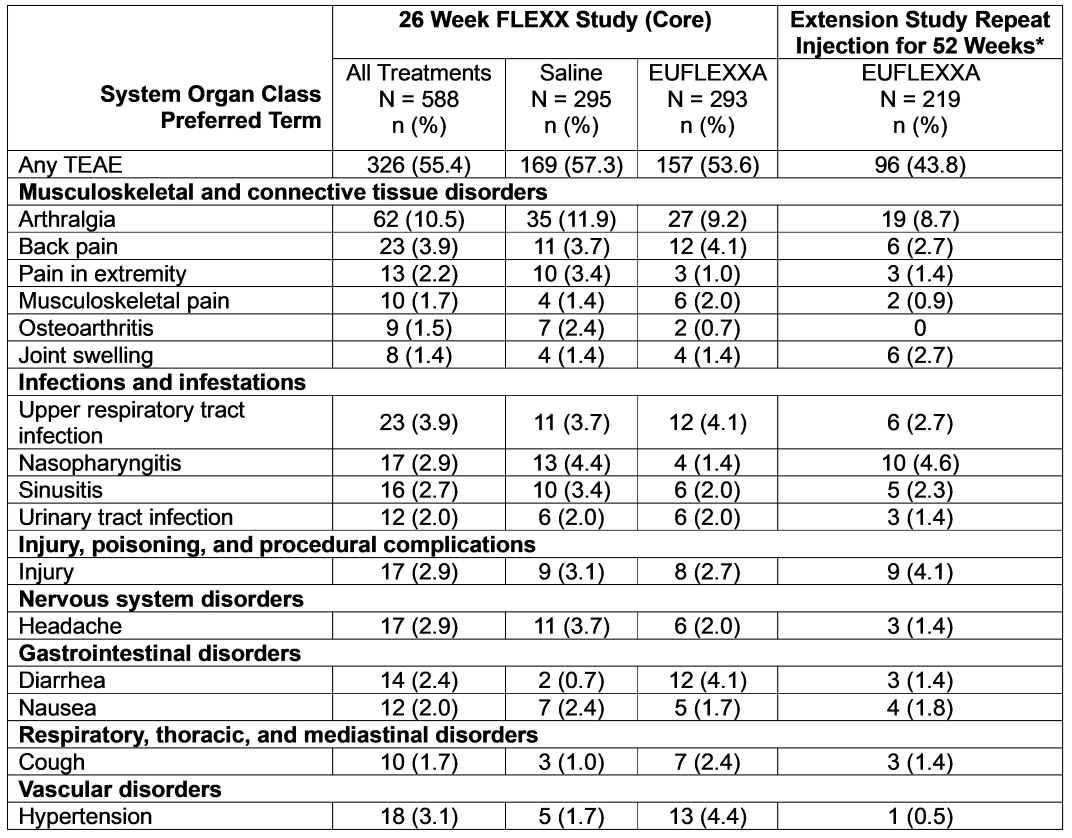
This was a multicenter, randomized, double-blind trial evaluating the efficacy and safety of EUFLEXXA, as compared with saline, in subjects with chronic osteoarthritis of the knee followed by an open labeled safety extension study. The intervention consisted of three (3) weekly injections of study device into the target knee, with scheduled follow-up evaluations during the 26 weeks following the first injection. In the extension phase subjects received three (3) weekly injections of EUFLEXXA into the target knee with follow-up evaluation up to 52 weeks. Table 3 shows the treatment-emergent adverse events by preferred term with an incidence of ≥2% among treatment groups.
*Treatment group for repeat study are for subjects who received EUFLEXXA in both the core and extension (219 out of 433). N = number of subjects in a given treatment group for the population analyzed; n = number of subjects reporting at least one adverse event within system organ class/preferred term; (%) = percentage of subjects based on N; TEAE = treatment- emergent adverse event. Note: An adverse event was counted as a TEAE if it was either not present at baseline (prior to the first dose of double-blind study device) or present at baseline but increased in severity during the treatment period.
During the initial randomization/treatment phase, 326 (55.4%) subjects in the safety population experienced 742 TEAEs. The proportion of subjects reporting TEAEs was generally similar in the EUFLEXXA and saline groups (53.6% and 57.3%, respectively). The most common preferred term of TEAE was arthralgia (10.5% of all subjects). Thirty (5.1%) subjects experienced severe TEAEs, and the proportion with severe events was larger in the saline group (6.4%) than the EUFLEXXA group (3.8%). Overall, 10.4% of subjects had TEAEs considered related to study device, with comparable proportions in each treatment group (9.9% and 10.8% for EUFLEXXA and saline, respectively).
During the extension phase, 43.4% (188/433) of subjects reported 377 TEAEs. Of these 43.8% (96/219) subjects receiving repeated EUFLEXXA reported 199 TEAEs. The most frequently reported preferred term in subjects formerly assigned to the core study EUFLEXXA group were arthralgia (8.7%), nasopharyngitis (4.6%), injury (4.1%), upper respiratory tract infections (2.7%), joint swelling (2.7%), back pain (2.7%), and sinusitis (2.3%). Of these TEAEs 9 (4.1%) subjects had study device related AEs classified as “Certain,” “Probable,” “Possible” or “Un-assessable.” The most common related TEAEs were arthralgia (2.3%) and joint swelling (1.4%). Table 4 shows the Study Device Related Treatment-Emergent Adverse Events by Preferred Term with an Incidence of ≥1 among Treatment Groups (Safety Population).
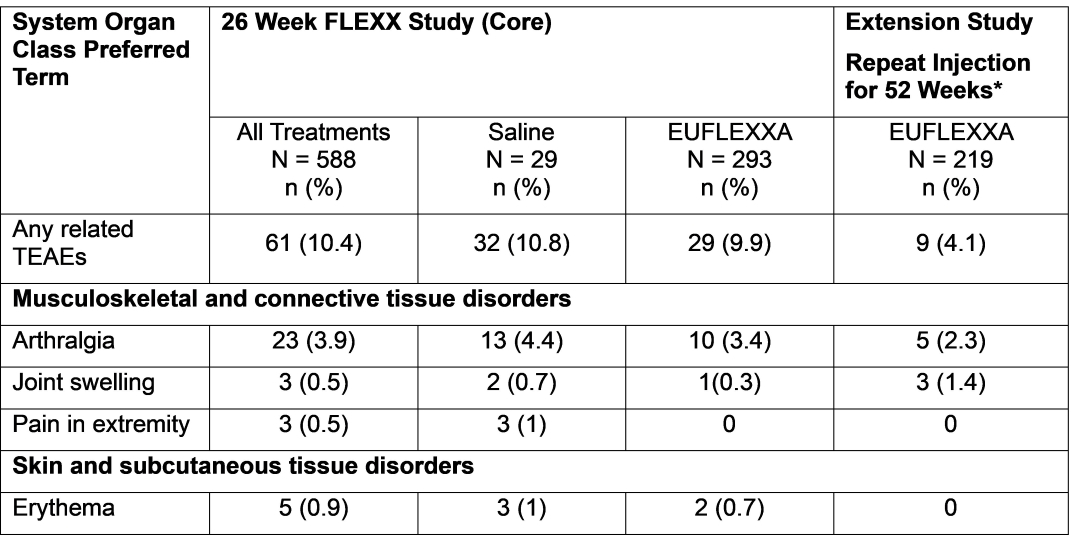
*TEAEs are for subjects who received EUFLEXXA in both the core and extension (219 out of 433).
N = number of subjects in a given treatment group for the population analyzed; n = number of subjects reporting at least 1 AE within system organ class/preferred term; (%) = percentage of subjects based on N; TEAE = treatment-emergent adverse event.
Note: Related AEs are AEs with study device relationship classified as “Certain,” “Probable,” “Possible” or “Un-assessable.”
Twenty-three serious TEAEs were reported in 19 (3.2%) subjects during the study: 10 (3.4%) subjects in the EUFLEXXA group and 9 (3.1%) subjects in the saline group. One of these events was considered related to the study device (increased redness of the left knee joint in the EUFLEXXA group). Eight (1.4%) subjects had 9 TEAEs leading to discontinuation: 3 (1.0%) subjects in the EUFLEXXA group and 5 (1.7%) subjects in the saline group.
Twelve (2.8%) subjects reported 20 serious TEAEs during the extension phase. Six of these subjects had received EUFLEXXA during the core study. None of the serious TEAEs was considered related to study device, and all resolved. Two (0.5%) subjects had TEAEs leading to discontinuation from the study, one of whom received EUFLEXXA during the core study; both subjects had events that were considered unrelated to study device.
Two subjects on saline experienced joint effusion. There were no reports of joint effusion among subjects receiving EUFLEXXA during the core and extension phase.
The safety and effectiveness of EUFLEXXA as a treatment for pain in OA of the knee was investigated in a multicenter clinical trial conducted in Germany.
The clinical investigation was a prospective randomized, double-blinded, active control (commercially available hyaluronan) study conducted at 10 centers in Germany. A total of 321 patients with stage 2 – 3 osteoarthritis of the knee according to the Kellgren and Lawrence grading system, meeting the Altman Criteria for Classification of Idiopathic Osteoarthritis of the knee, and scoring an average score of 41 – 80 mm on the WOMAC VAS pain index were randomized into groups of equal size to receive either EUFLEXXA (160 patients) or the active control (161 patients).
The demographics of trial participants were comparable across treatment groups with regard to age, gender, Kellgren and Lawrence grading system, stiffness, crepitus, bony enlargement, and no palpable warmth. Table 5 lists the demographics of the patient population.
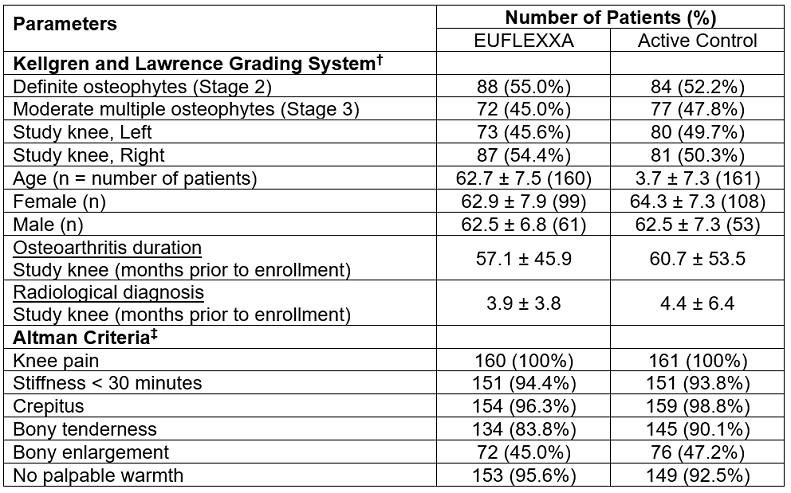
† Kellgren and Lawrence (Ann Rheum Dis 1957;(16):494-501): Based on radiological findings, osteoarthritis stages were defined as follows: 0 = normal, 1 = doubtful narrowing of joint space and possible osteophytic lipping, 2 = definite osteophytes and possible narrowing of joint space, 3 = moderate multiple osteophytes and definite narrowing of joint space, some sclerosis and possible deformity of bone contour, 4 = large osteophytes, marked narrowing of joint space, severe sclerosis and definite deformity of bone contour.
‡ Altman, et al., (Arthritis and Rheumatism 1986;29(8):1039-1049): Clinical criteria for classification of idiopathic osteoarthritis (OA) of the knee were defined as follows: Knee pain and at least 3 of the following 6 parameters: Age > 50 years, Stiffness < 30 minutes, Crepitus, Bony tenderness, Bony enlargement, No palpable warmth.
Clinical Results
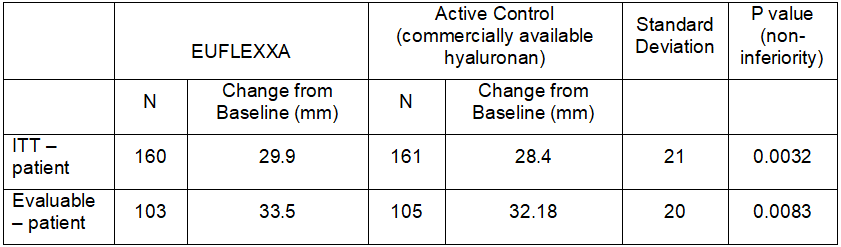
For this trial, the main performance analysis for determining non-inferiority was determined using the improvement in the average of the five patient’s self-evaluation pain parameters measured by the VAS WOMAC index at Week 12 from baseline. This analysis was performed for both the intent-to-treat population, (i.e., every subject who received the injection), and the evaluable population, (i.e., those subjects who had average pain scores of 41-80 allowing only one parameter to be below 20 or above 80 at both the pre-screening visit and visit 1). For those patients who dropped out of the study before Week 12, the last evaluation was used. For those patients who requested NSAID or analgesic during the study, the last evaluation before start of NSAID/analgesic was used for the analysis. The results indicate that the effect of EUFLEXXA on pain relief was not inferior to that of a commercially available hyaluronan.
26 Week Multicenter Clinical Trial
This was a multicenter, randomized, double-blind trial evaluating the efficacy and safety of EUFLEXXA as compared to saline comparator in subjects with chronic osteoarthritis of the knee. The intervention consisted of three weekly injections into the target knee with evaluations from baseline through Week 26 (1, 2, 3, 6, 12, 18, and 26). The primary objective was to demonstrate superiority over saline comparator from baseline to Week 26 using the pain level reported following a 50 foot walk test, measured by 100 mm visual analog scale. The following secondary endpoints were also evaluated: OARSI responder rate at Week 12 and Week 26; WOMAC pain, disability, and joint stiffness score changes from baseline to Week 12 and 26; and change in Patient Global Assessment from baseline to Week 12 and Week 26.
Patient Population and Demographics
A total of 821 subjects were screened for the study, and 588 subjects were randomized. Approximately 88% of the randomized subjects completed the study, with similar proportions completing in each treatment group. Sixty-eight (11.6%) subjects discontinued the randomization/treatment phase prematurely: 34 (11.5%) in the saline group and 34 (11.6%) in the EUFLEXXA group. The most common reasons for discontinuation were the subject’s withdrawing consent 25 (4.3%) and AEs 17 (2.9%). A total of 433 (73.6%) subjects entered the open-label extension study.
Clinical Results
Primary Endpoint
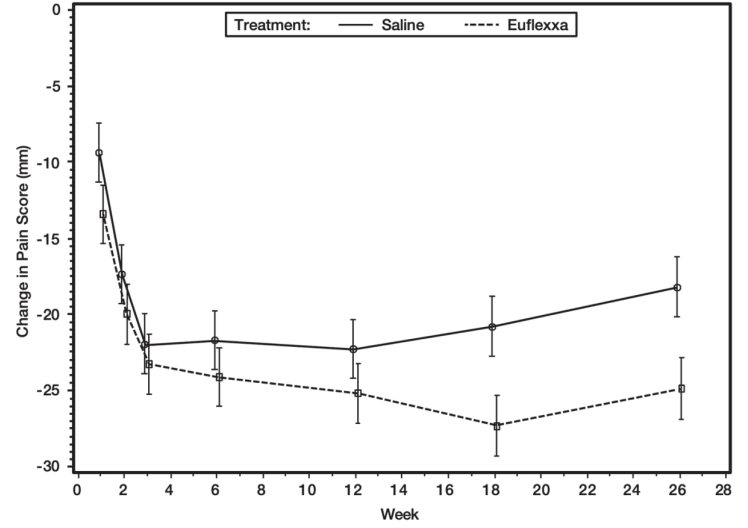
In the primary efficacy analysis, the EUFLEXXA group showed a larger mean decrease in pain scores on the 50-foot walk test from baseline to Week 26 than the saline group: –25.7 (28.85) mm versus –18.5 (32.53) mm, respectively. The group difference in least squares mean change from baseline of -6.6 mm (95% CI = –10.8 to –2.5 mm) was statistically significant (p-value = 0.002). Figure 1 depicts the adjusted mean change in pain scores on 50-foot walk test from baseline to week 26 (ITT Population).
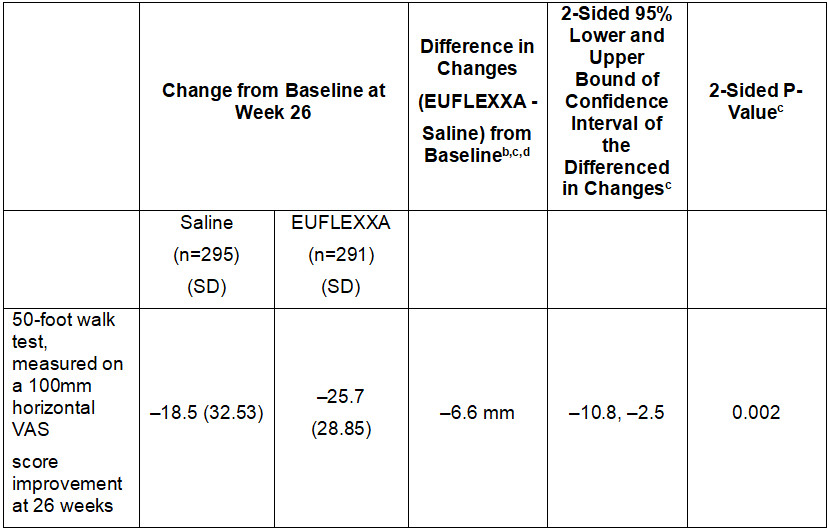
aITT= Intent to Treat.
bNegative (–) values favor EUFLEXXA.
cThe analysis is based on repeated measure mixed model Analysis of Covariance (ANCOVA) from baseline through 26 weeks on mean change from baseline 50-foot walk test, measured on a 100mm horizontal VAS score improvement at 26 weeks, with a weekly injection of EUFLEXXA for 3 weeks.
ddifference = least squares mean difference.
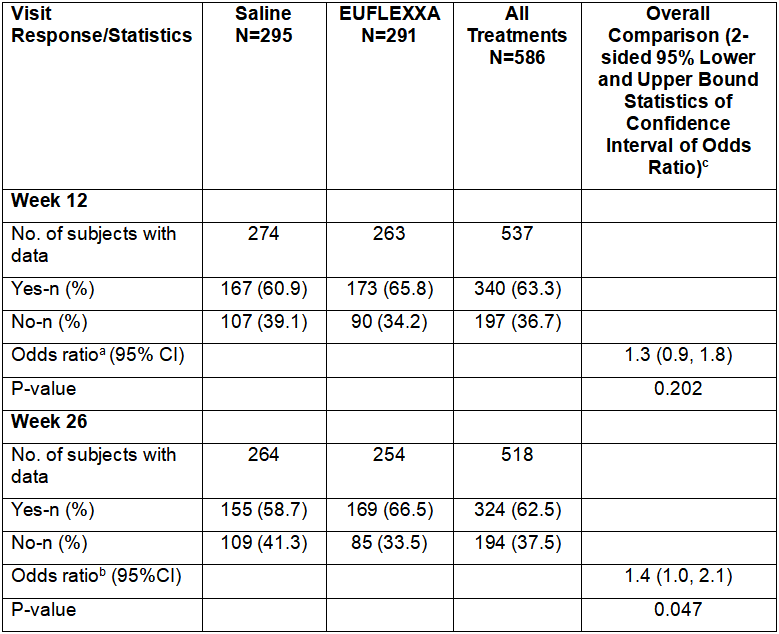
OARSI = Osteoarthritis Research Society International; ITT = intent-to-treat; N = number of subjects in a given treatment group for the population analyzed; n = number of subjects; (%) = percentage of subjects based on N; CI = confidence interval.
Note: The p-value for the odds ratio corresponds to the Wald chi-square test for EUFLEXXA versus saline with respect to OARSI responder rates from a logistic regression adjusting for treatment group and study center.
Note: A subject was considered a responder if there was high improvement in pain or function >50% and absolute change >20 mm or improvement in at least two of the three following categories: pain >20% and absolute change >10 mm, function >20% and absolute change >10 mm, and/or Patient Global Assessment >20% and absolute change >10.
a,be (Log Odds Ratio)= 1.27 for 12 weeks and 1.4 for 26 weeks, based on a logistic regression model. (Log Odds Ratio)=loge[probability (responder)/probability (non-responder)] EUFLEXXA / [probability (responder)/ probability(non-responder)]saline.
cWhen odds ratio >1, [probability(responder)/probability (non-responder)] EUFLEXXA > [probability (responder)/probability (nonresponder)saline].
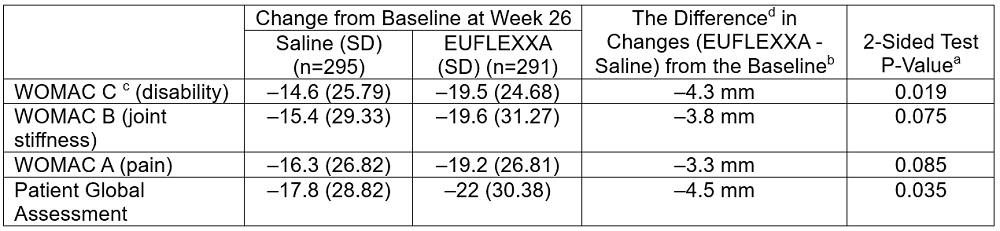
Note: The analysis is based on repeated measure mixed model Analysis of Covariance (ANCOVA) from baseline through 26 weeks on mean change from baseline.
aP-values are not adjusted for the multiplicity.
bNegative (–) values for WOMAC C and Patient Global Assessment are in favor of EUFLEXXA.
c The Western Ontario and McMaster Universities Arthritis Index (WOMAC) is a set of standardized questionnaires used by healthcare professionals to evaluate the condition of patients with osteoarthritis of the knee and hip. WOMAC Pain Scale is 100mm.
ddifference=least square mean difference.
No significant treatment group differences were observed in the change in number of study-specific acetaminophen tablets used per week or in the proportion of subjects who were pain free at Week 26 or last visit.
EUFLEXXA is supplied in 2.25 mL nominal volume, disposable, pre-filled glass syringes containing 2 mL of EUFLEXXA. Only the contents of the syringe are sterile. EUFLEXXA is nonpyrogenic.
This product is not made with natural rubber latex. Product Number: 55566-4100-1.
3 disposable syringes per carton.
Federal law restricts this device to sale by or on the order of a physician.
EUFLEXXA® (1% sodium hyaluronate)
Be sure to read the following important information carefully.
This information does not take the place of your doctor’s advice. If you do not understand this information or want to know more, ask your doctor.
EUFLEXXA is a gel-like, elastic, sterile product containing natural, highly purified hyaluronan (pronounced hye-a-loo-ROE-nan). Hyaluronan is a natural substance found in the body. It is present in particularly high amounts in joint tissues and in the fluid that fills the joints. The body’s own hyaluronan acts like a lubricant and a shock absorber in the joint. It is needed for the joint to function properly.
Unlike some other hyaluronan products, EUFLEXXA does not come from bird- derived sources.
EUFLEXXA is used to relieve knee pain due to osteoarthritis (pronounced Os-tee-oh-are-THRY-tis). It is used for patients who do not get enough relief from simple pain medications such as acetaminophen or from exercise and physical therapy.
Osteoarthritis is a condition that involves the wearing down of cartilage (the protective covering on the ends of your bones). In osteoarthritis, there may not be enough or a decrease in the quality of the gel-like substance in the joint and surrounding tissues.
EUFLEXXA comes in pre-filled syringes containing 2 mL (about half a teaspoon) of product. EUFLEXXA is given by injection directly into the knee joint by a doctor or other qualified healthcare professional. EUFLEXXA is injected into your knee once a week, for a total of three injections.
Your doctor will determine if you are not an appropriate candidate for EUFLEXXA.
The following is a list of adverse effects that can occur with either EUFLEXXA or saline injections.
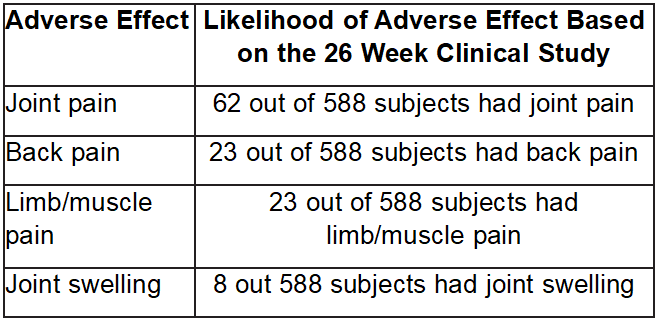
If you have a reaction where the swelling is extensive and painful, you should call your doctor.
Based on the clinical studies, EUFLEXXA subjects had pain relief and improvement in function lasting up to 6 months.
– Running
– Tennis
– Hiking
– Jumping
– Swimming
– Heavy lifting (weight lifting)
– Jogging
– Bicycling
– Aerobic exercise
If you experience any of the adverse effects or symptoms described earlier or if you have any other problems, you should call your doctor immediately.
If you have osteoarthritis, there are other non-surgical treatment options available and these include:
A medical study involving 321 patients with knee pain due to osteoarthritis was performed in Germany. The study compared EUFLEXXA against another hyaluronan once a week for 3 weeks (control arm).
Pain, stiffness and function of the knee joint and patients’ and doctors’ judgment of treatment success were measured for 12 weeks. Patients were those with knee paindue to osteoarthritis who had not received pain relief with other medications. Patients experienced pain relief from EUFLEXXA injections similar to those patients in the control arm.
Another study involving 588 patients with knee pain due to osteoarthritis was conducted in the United States. Two hundred ninety three (293) patients were injected with EUFLEXXA and 295 with saline (salt water). The pain scores were used to compare the effectiveness of EUFLEXXA to saline injection: Patients were asked to rate how pain was felt on the 100 mm scale after 50 foot walk at 1, 2, 3, 6, 12, 18 and 26 weeks. EUFLEXXA group improved 25.7 mm from the baseline pain score, whereas the saline group improved 18.5 mm. There was more improve- ment in EUFLEXXA group than the saline group. The difference was 6.6 mm on 100 mm pain scale in favor of EUFLEXXA group. Study results showed significant improvement in osteoarthritis knee pain relief with EUFLEXXA therapy lasting up to 6 months. The study also showed that a repeated cycle of EUFLEXXA for an additional 26 weeks (1 year total) was safe.
EUFLEXXA has not been proven to relieve pain in any other joints.
The number of subjects reporting adverse events was generally similar between the EUFLEXXA and Saline groups. Serious events were not observed in these clinical studies.
The following are the most common adverse events and symptoms that occurred during clinical studies of EUFLEXXA:
If you have any questions or problems, talk to your doctor. If you would like more information on EUFLEXXA, please call 1-888-FERRING (1-888-337-7464) toll-free or visit www.euflexxa.com.
MANUFACTURED FOR:
![]()
FERRING PHARMACEUTICALS INC. PARSIPPANY, NJ 07054
6309501103
Rev. 07/2016
EUFLEXXA is a registered trademark of Ferring BV


