


OUR MEDICINES
A range of medicines have been developed by Ferring in core therapy areas.
Which medicines have been developed?
Which medicines have been developed?


FIRMAGON® is a GnRH receptor antagonist indicated for treatment of patients with advanced prostate cancer. It is intended to be administered subcutaneously into the patient’s abdomen by a trained healthcare professional (HCP).
FIRMAGON® was approved by the FDA in 2008.
Below are the key scientific publications, which contain the data that substantiate the FDA approval of FIRMAGON® (degarelix) powder.
This product label was last updated on Feb - 2020
Warnings and Precautions, Embryo-Fetal Toxicity (5.4)
FIRMAGON is a GnRH receptor antagonist indicated for treatment of patients with advanced prostate cancer. (1)
For injection:
Most common adverse reactions (≥10%) are injection site reactions (e.g., pain, erythema, swelling or induration), hot flashes, and increases in serum levels of transaminases and gamma- glutamyltransferase (GGT) (6.1)
*Sections or subsections omitted from the full prescribing information are not listed.
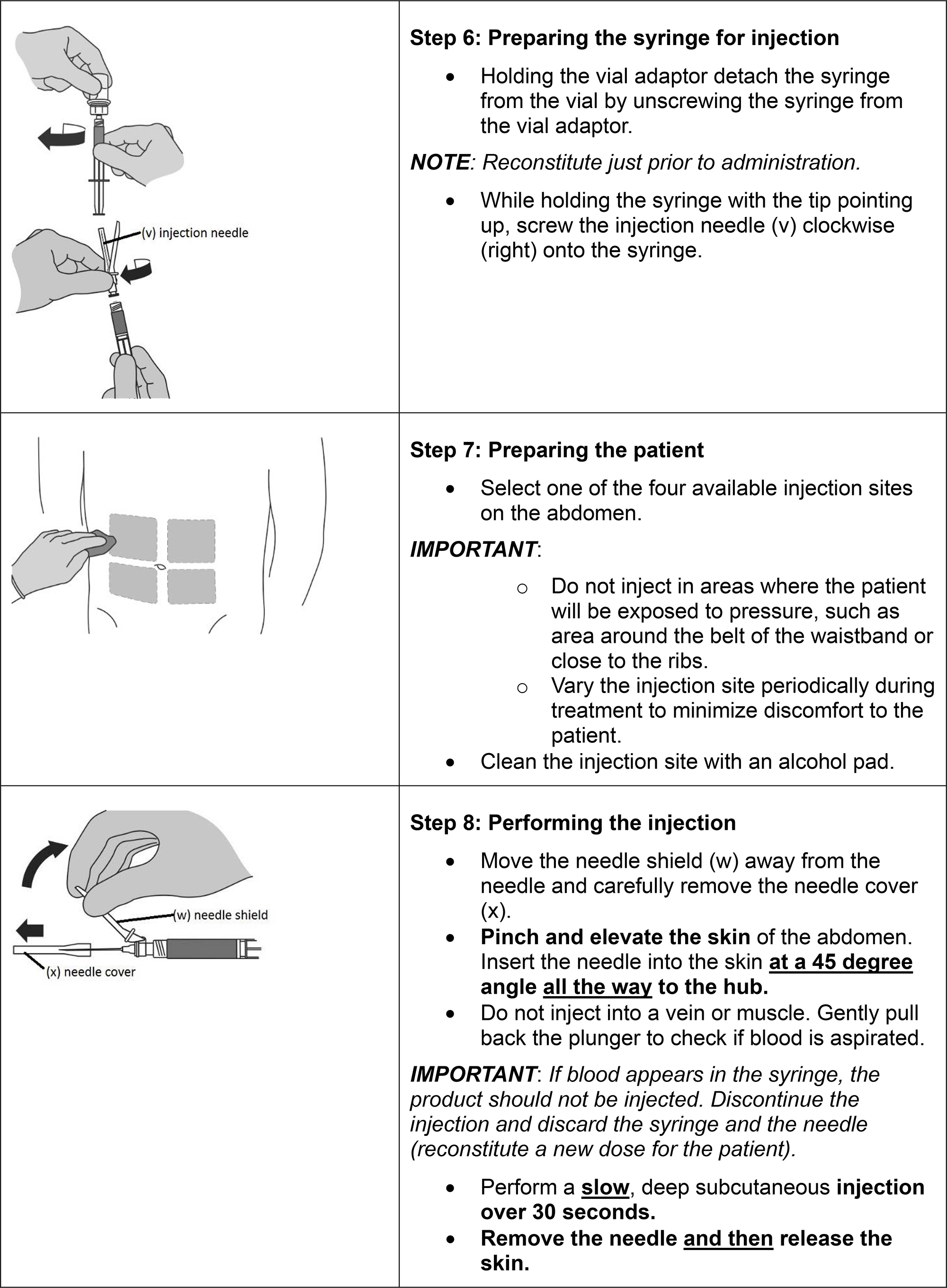
FIRMAGON is administered as a subcutaneous injection in the abdominal region only at the dosages in Table 1 below.
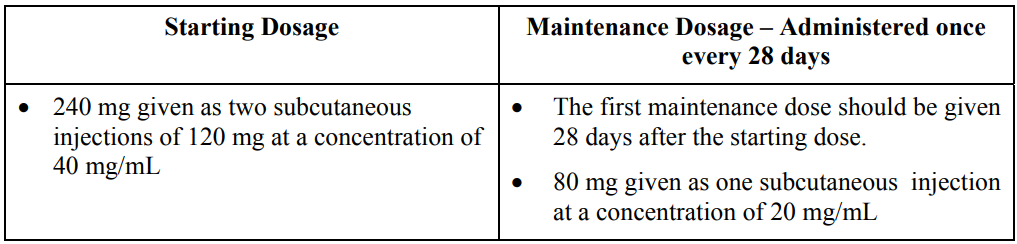
FIRMAGON is to be administered by a healthcare professional only.
Before administering FIRMAGON read the Instructions for reconstitution and administration carefully. As with other drugs administered by subcutaneous injection, the injection site should vary periodically. Injections should be given only in areas of the abdomen that will not be exposed to pressure, e.g., not close to waistband or belt nor close to the ribs.
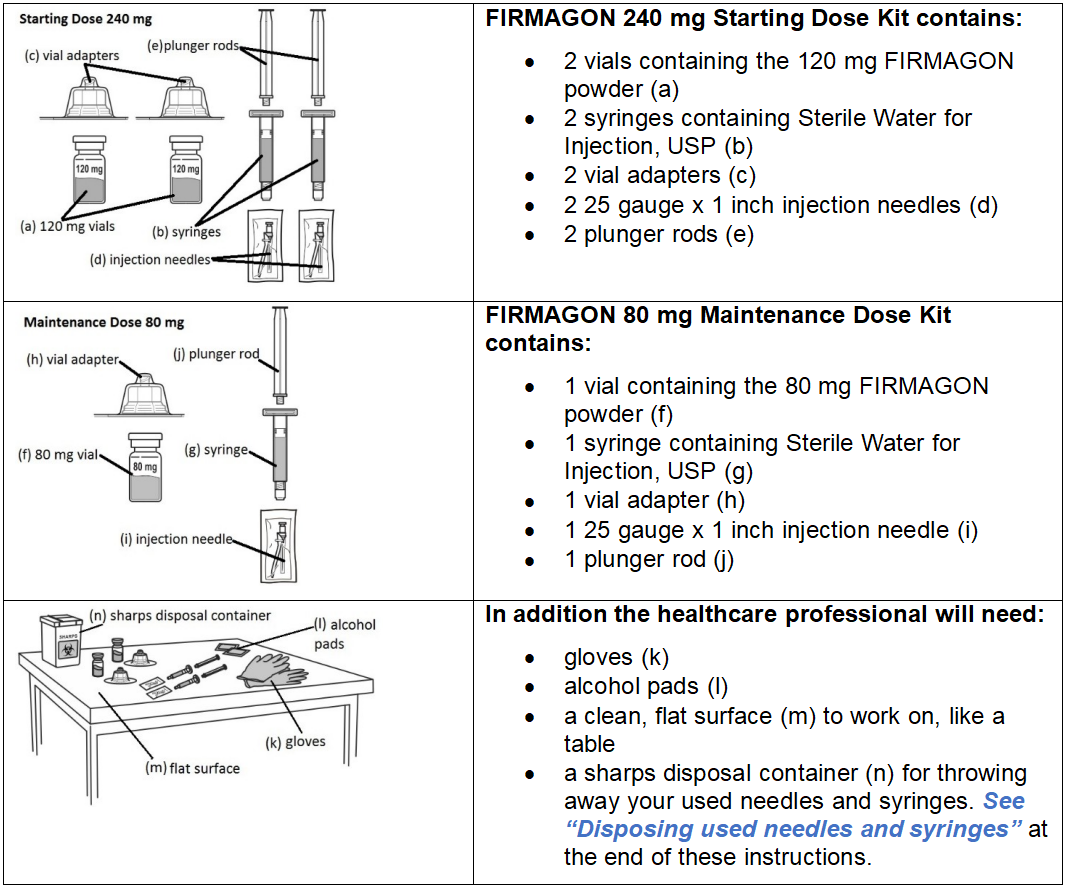
FIRMAGON is supplied as a powder to be reconstituted with Sterile Water for Injection, USP.
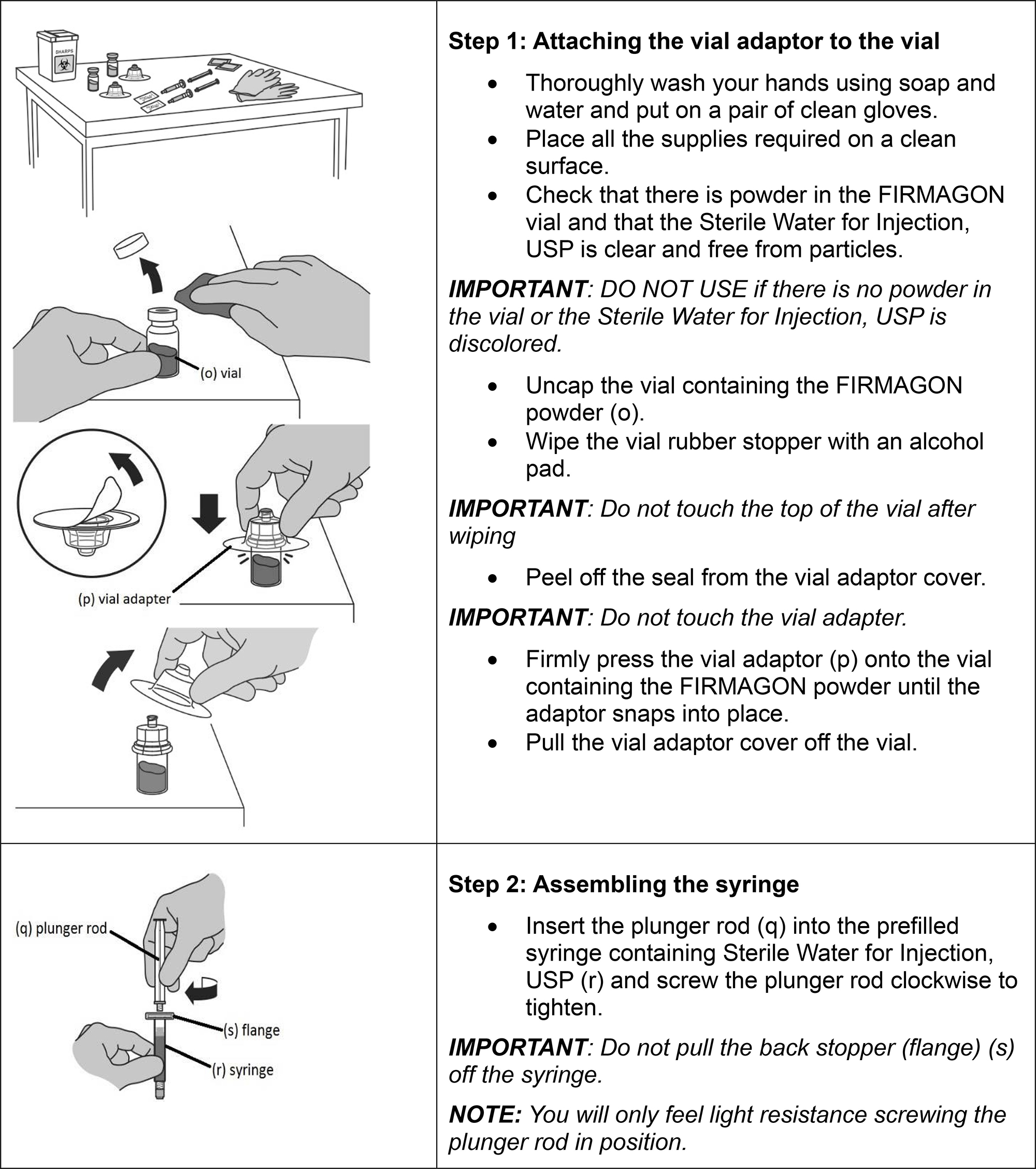
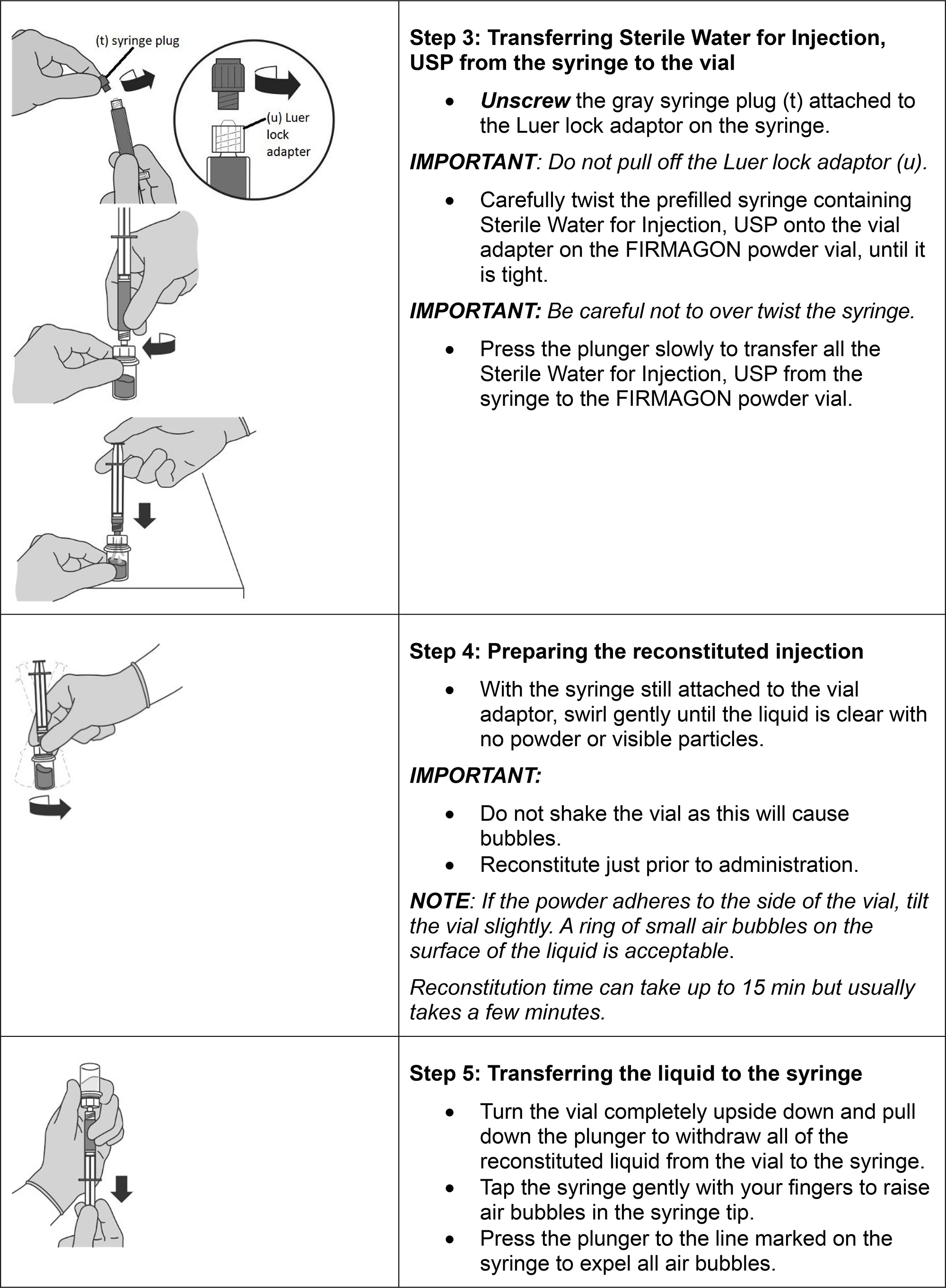
Follow the instructions for reconstitution closely and read the complete instructions before performing the subcutaneous injection.
Reconstituted drug must be administered within one hour after addition of Sterile Water for Injection, USP.
Do not shake the vials.
Follow aseptic technique.
NOTE: The mixing process must be repeated for the two injections of the Starting Dose prior to injecting the product into the patient’s abdomen.
For injection:
FIRMAGON is contraindicated in patients with history of severe hypersensitivity to degarelix or to any of the product components [see Warnings and Precautions (5.1)].
FIRMAGON is contraindicated in patients with history of severe hypersensitivity to degarelix or to any of the product components [see Contraindications (4)].
Hypersensitivity reactions, including anaphylaxis, urticaria and angioedema, have been reported post-marketing with FIRMAGON.
In case of a severe hypersensitivity reaction, discontinue FIRMAGON immediately if the injection has not been completed, and manage as clinically indicated. Patients with a known history of severe hypersensitivity reactions to FIRMAGON should not be re-challenged with FIRMAGON
Androgen deprivation therapy may prolong the QT interval. Providers should consider whether the benefits of androgen deprivation therapy outweigh the potential risks in patients with congenital long QT syndrome, congestive heart failure, frequent electrolyte abnormalities, and in patients taking drugs known to prolong the QT interval. Electrolyte abnormalities should be corrected. Consider periodic monitoring of electrocardiograms and electrolytes.
In the randomized, active-controlled trial comparing FIRMAGON to leuprolide, periodic electro-cardiograms were performed. Seven patients, three (<1%) in the pooled degarelix group and four (2%) patients in the leuprolide 7.5 mg group, had a QTcF ≥500 msec. From baseline to end of study, the median change for FIRMAGON was 12.3 msec and for leuprolide was 16.7 msec.
FIRMAGON results in suppression of the pituitary gonadal system. Results of diagnostic tests of the pituitary gonadotropic and gonadal functions conducted during and after FIRMAGON may be affected. The therapeutic effect of FIRMAGON should be monitored by measuring serum concentrations of prostate-specific antigen (PSA) periodically. If PSA increases, serum concentrations of testosterone should be measured.
Based on findings in animal studies, FIRMAGON can cause fetal harm and loss of pregnancy when administered to a pregnant woman. In animal developmental and reproductive toxicity studies in rats and rabbits, oral administration of degarelix during organogenesis caused embryo-fetal lethality and abortion as well as increased post-implantation loss and decreased the number of live fetuses in animals at doses less than the clinical loading dose based on body surface area. Advise pregnant patients and females of reproductive potential of the potential risk to the fetus [see Use in Specific Populations (8.1)].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
FIRMAGON was studied in a randomized, open-label trial in which patients with prostate cancer were randomized to receive FIRMAGON (subcutaneous) or leuprolide (intramuscular) monthly for 12 months [see Clinical Studies (14)].
The most common adverse reactions (≥10%) during FIRMAGON therapy are injection site reactions (e.g., pain, erythema, swelling or induration), hot flashes, and increases in serum levels of transaminases and gammaglutamyltransferase (GGT). The majority of the adverse reactions were Grade 1 or 2, with Grade 3/4 adverse reaction incidences of 1% or less.
Adverse reactions reported in ≥ 5% of patients treated with FIRMAGON (subcutaneous) 240 mg starting dose and then 80 mg maintenance dose once every 28 days or who were treated with 7.5 mg of leuprolide (intramuscular) every 28 days are shown in Table 2.
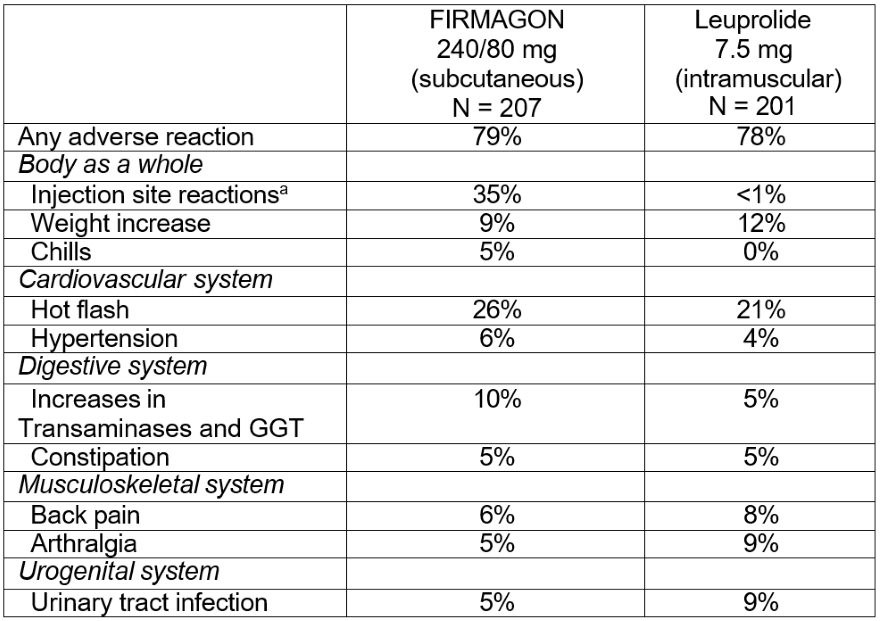
a Includes pain, erythema, swelling, induration, or nodule.
The following adverse reactions occurred in 1 to < 5% of patients treated with FIRMAGON:
Body as a whole: Asthenia, fatigue, fever, night sweats
Digestive system: Nausea
Nervous system: Dizziness, headache, insomnia
The following adverse reactions, not already listed, occurred in ≥ 1% of patients treated in any study with FIRMAGON:
Reproductive System: Erectile dysfunction, testicular atrophy
Endocrine Disorders: Gynecomastia General: Hyperhidrosis Gastrointestinal: Diarrhea
Injection Site Reactions
The most frequently reported adverse reactions at the injection sites were pain (28%), erythema (17%), swelling (6%), induration (4%) and nodule (3%). These adverse reactions were mostly transient, of mild to moderate intensity, occurred primarily with the starting dose and led to few discontinuations (<1%). Grade 3 injection site reactions occurred in 2% or less of patients receiving FIRMAGON.
Hepatic Laboratory Abnormalities
Hepatic laboratory abnormalities were primarily Grade 1 or 2 and were generally reversible. Grade 3 hepatic laboratory abnormalities occurred in less than 1% of patients.
FIRMAGON Extension Study
The safety of FIRMAGON administered once every 28 days was evaluated further in an extension study (NCT00451958) in 385 patients who completed the above active-controlled trial. Of the 385 patients, 251 patients continued treatment with FIRMAGON and 135 patients crossed over treatment from leuprolide to FIRMAGON.
The median treatment duration on the extension study was approximately 43 months (range 1 to 58 months). The most common adverse reactions reported in >10% of the patients were injection site reactions (e.g., pain, erythema, swelling, induration or inflammation), pyrexia, hot flush, weight loss or gain, fatigue, increases in serum levels of hepatic transaminases and GGT. One percent of patients had injection site infections including abscess. Hepatic laboratory abnormalities in the extension study included the following: Grade 1/2 elevations in hepatic transaminases occurred in 47% of patients and Grade 3 elevations occurred in 1% of patients.
As with all peptides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease.
Anti-Degarelix Antibodies
Anti-degarelix antibody development has been observed in 10% of patients after treatment with FIRMAGON for 1 year. There is no indication that the efficacy or safety of FIRMAGON treatment is affected by antibody formation.
The following adverse reactions have been identified during post-approval use of FIRMAGON. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Changes in bone density
Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with a GnRH agonist. It can be anticipated that long periods of medical castration in men will result in decreased bone density.
No drug-drug interaction studies were conducted.
Degarelix is not a substrate for the human CYP450 system. Degarelix is not an inducer or inhibitor of the CYP450 system in vitro. Therefore, clinically significant CYP450 pharmacokinetic drug-drug interactions are unlikely.
Risk Summary
The safety and efficacy of FIRMAGON have not been established in women.
Based on findings in animal studies and mechanism of action, FIRMAGON can cause fetal harm and loss of pregnancy when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no human data on the use of FIRMAGON in pregnant women to inform the drug-associated risk. In animal developmental and reproductive toxicity studies in rats and rabbits, oral administration of degarelix during organogenesis caused embryo-fetal lethality and abortion as well as increased post-implantation loss and decreased the number of live fetuses in animals at doses less than the clinical loading dose based on body surface area (see Data). Advise pregnant patients and females of reproductive potential of the potential risk to the fetus.
Data
Animal Data
When degarelix was given to rabbits during early organogenesis at doses of 0.002 mg/kg/day (about 0.02% of the clinical loading dose based on body surface area), there was an increase in early post-implantation loss. Degarelix given to rabbits during mid and late organogenesis at doses of 0.006 mg/kg/day (about 0.05% of the clinical loading dose based on body surface area) caused embryo/fetal lethality and abortion. When degarelix was given to female rats during early organogenesis, at doses of 0.0045 mg/kg/day (about 0.036% of the clinical loading dose based on body surface area), there was an increase in early post-implantation loss. When degarelix was given to female rats during mid and late organogenesis, at doses of 0.045 mg/kg/day (about 0.36% of the clinical loading dose based on body surface area), there was an increase in the number of minor skeletal abnormalities and variants.
The safety and efficacy of FIRMAGON have not been established in females. There are no data on the presence of degarelix in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are present in human milk and because of the potential for serious adverse reactions in a breastfed child from degarelix, a decision should be made whether to discontinue nursing or discontinue the drug taking into account the importance of the drug to the mother.
Infertility
Based on findings in animals and mechanism of action, degarelix may impair fertility in males and females of reproductive potential [see Nonclinical Toxicology (13.1)].
Safety and effectiveness in pediatric patients have not been established.
Of the total number of subjects in clinical studies of FIRMAGON, 82% were age 65 and over, while 42% were age 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
No pharmacokinetic studies in renally impaired patients have been conducted. At least 20-30% of a given dose of degarelix is excreted unchanged in the urine. A population pharmacokinetic analysis of data from the randomized study demonstrated that there is no significant effect of mild renal impairment [creatinine clearance (CrCL) 50–80 mL/min] on either the degarelix concentration or testosterone concentration. Data on patients with moderate or severe renal impairment is limited and therefore degarelix should be used with caution in patients with CrCL <50 mL/min.
Patients with hepatic impairment were excluded from the randomized trial.
A single dose of 1 mg degarelix administered as an intravenous infusion over 1 hour was studied in 16 non-prostate cancer patients with either mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. Compared to non-prostate cancer patients with normal liver function, the exposure of degarelix decreased by 10% and 18% in patients with mild and moderate hepatic impairment, respectively. Therefore, dose adjustment is not necessary in patients with mild or moderate hepatic impairment. However, since hepatic impairment can lower degarelix exposure, it is recommended that in patients with hepatic impairment testosterone concentrations should be monitored on a monthly basis until medical castration is achieved. Once medical castration is achieved, an every- other-month testosterone monitoring approach could be considered.
Patients with severe hepatic dysfunction have not been studied and caution is therefore warranted in this group.
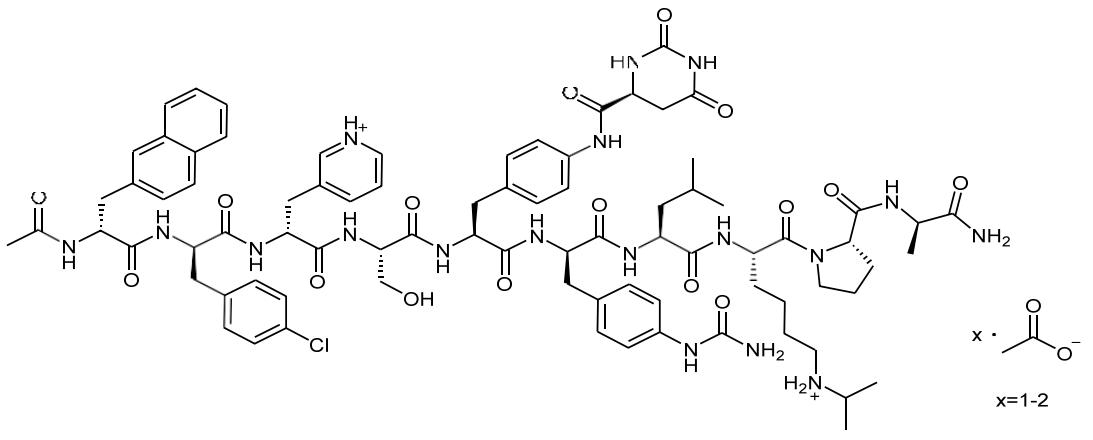
FIRMAGON is a sterile lyophilized powder for injection containing degarelix (as the acetate) and mannitol. Degarelix is a synthetic linear decapeptide amide containing seven unnatural amino acids, five of which are D- amino acids. The acetate salt of degarelix is a white to off-white amorphous powder of low density as obtained after lyophilization.
The chemical name of degarelix is D-Alaninamide, N-acetyl-3-(2-naphthalenyl)-D-alanyl-4-chloro-D- phenylalanyl-3-(3-pyridinyl)-D-alanyl-L-seryl-4-[[[(4S)-hexahydro-2,6-dioxo-4-pyrimidinyl]carbonyl]amino]-L phenylalanyl-4-[(aminocarbonyl)amino]-D-phenylalanyl-L-leucyl-N6–(1-methylethyl)-L-lysyl-L-prolyl. It has an empirical formula of C82H103N18O16Cl and a molecular weight of 1632.3 Da.
Degarelix acetate has the following structural formula:
FIRMAGON is available in two packaging configurations:
Degarelix is a GnRH receptor antagonist. It binds reversibly to the pituitary GnRH receptors, thereby reducing the release of gonadotropins and consequently testosterone.
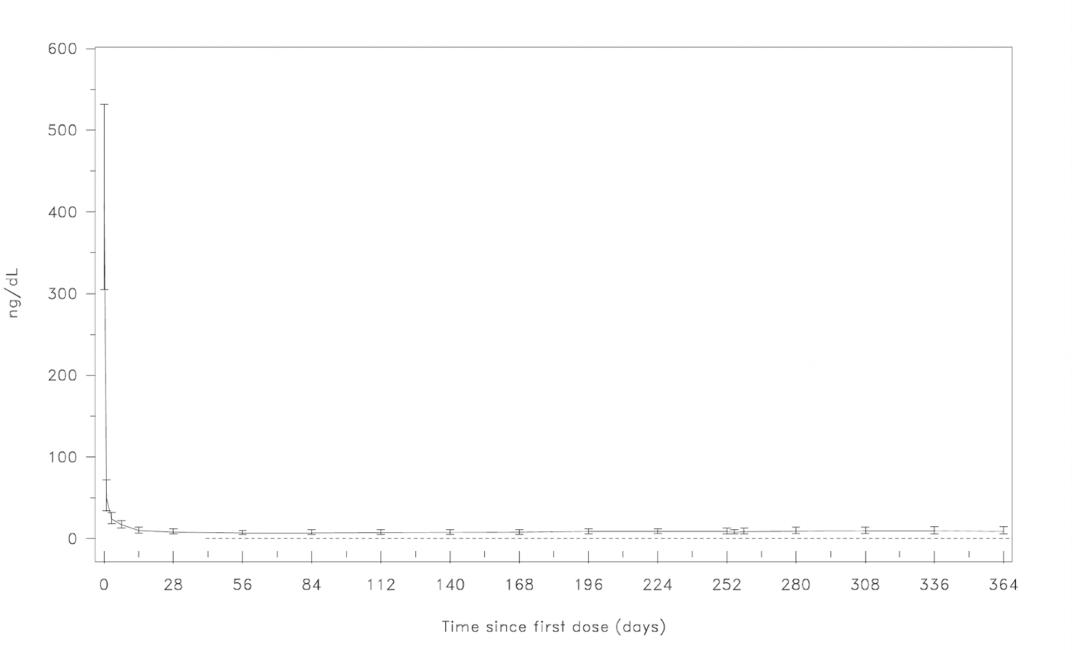
A single dose of 240 mg FIRMAGON causes a decrease in the plasma concentrations of luteinizing hormone (LH) and follicle stimulating hormone (FSH), and subsequently testosterone.
FIRMAGON is effective in achieving and maintaining testosterone suppression below the castration level of 50 ng/dL.
Absorption
FIRMAGON forms a depot upon subcutaneous administration, from which degarelix is released to the circulation. Following administration of FIRMAGON 240 mg at a product concentration of 40 mg/mL, the mean Cmax was 26.2 ng/mL (coefficient of variation, CV 83%) and the mean AUC was 1054 ng·day/mL (CV 35%). Typically Cmax occurred within 2 days after subcutaneous administration. In prostate cancer patients at a product concentration of 40 mg/mL, the pharmacokinetics of degarelix were linear over a dose range of 120 to 240 mg. The pharmacokinetic behavior of the drug is strongly influenced by its concentration in the injection solution.
Distribution
The distribution volume of degarelix after intravenous (>1 L/kg) or subcutaneous administration (>1000L) indicates that degarelix is distributed throughout total body water. In vitro plasma protein binding of degarelix is estimated to be approximately 90%.
Metabolism
Degarelix is subject to peptide hydrolysis during the passage of the hepato-biliary system and is mainly excreted as peptide fragments in the feces. No quantitatively significant metabolites were detected in plasma samples after subcutaneous administration. In vitro studies have shown that degarelix is not a substrate, inducer or inhibitor of the CYP450 or p-glycoprotein transporter systems.
Excretion
Following subcutaneous administration of 240 mg FIRMAGON at a concentration of 40 mg/mL to prostate cancer patients, degarelix is eliminated in a biphasic fashion, with a median terminal half-life of approximately 53 days. The long half-life after subcutaneous administration is a consequence of a very slow release of degarelix from the FIRMAGON depot formed at the injection site(s). Approximately 20–30% of a given dose of degarelix was renally excreted, suggesting that approximately 70–80% is excreted via the hepato-biliary system in humans. Following subcutaneous administration of degarelix to prostate cancer patients the clearance is approximately 9 L/hr.
Effect of Age, Weight and Race
There was no effect of age, weight or race on the degarelix pharmacokinetic parameters or testosterone concentration.
Degarelix was administered subcutaneously to rats every 2 weeks for 2 years at doses of 2, 10 and 25 mg/kg (about 9, 45 and 120% of the recommended human loading dose on a mg/m2 basis). Long term treatment with degarelix at 25 mg/kg caused an increase in the combined incidence of benign hemangiomas plus malignant hemangiosarcomas in females.
Degarelix was administered subcutaneously to mice every 2 weeks for 2 years at doses of 2, 10 and 50 mg/kg (about 5, 22 and 120% of the recommended human loading dose (240 mg) on a mg/m2 basis). There was no statistically significant increase in tumor incidence associated with this treatment.
Degarelix did not cause genetic damage in standard in vitro assays (bacterial mutation, human lymphocyte chromosome aberration) nor in in vivo rodent bone marrow micronucleus tests.
Single degarelix doses of ≥ 1 mg/kg (about 5% of the clinical loading dose on a mg/m2 basis) caused reversible infertility in male rats. Single doses of ≥ 0.1 mg/kg (about 0.5% of the clinical loading dose on a mg/m2 basis) caused a decrease in fertility in female rats.
The safety and efficacy of FIRMAGON were evaluated in an open-label, multi-center, randomized, parallel-group study (NCT00295750) in patients with prostate cancer. A total of 620 patients were randomized to receive one of two FIRMAGON dosing regimens or leuprolide for one year:
FIRMAGON is not approved for use with monthly doses of 160 mg (40 mg/mL) subcutaneously.
Serum levels of testosterone were measured at screening, on Day 0, 1, 3, 7, 14, and 28 in the first month, and then monthly until the end of the study.
The clinical trial population (n=610) across all treatment arms had an overall median age of approximately 73 (range 50 to 98). The ethnic/racial distribution was 84% white, 6% black and 10% others. Disease stage was distributed approximately as follows: 20% metastatic, 29% locally advanced (T3/T4 Nx M0 or N1 M0), 31% localized (T1 or T2 N0 M0) and 20% classified as other (including patients whose disease metastatic status could not be determined definitively – or patients with PSA relapse after primary curative therapy). In addition, the median testosterone baseline value across treatment arms was approximately 400 ng/dL.
The primary objective was to demonstrate that FIRMAGON is effective achieving and maintaining testosterone suppression to castration levels (T ≤ 50 ng/dL) during 12 months of treatment. The results are shown in Table 3.
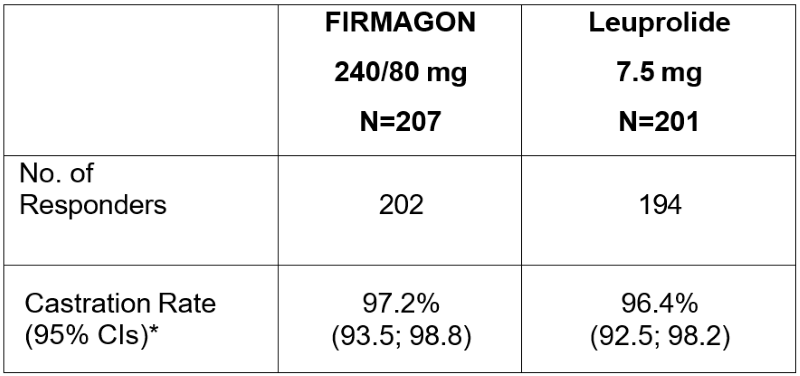
*Kaplan Meier estimates within group
Percentage changes in testosterone from baseline to Day 28 (median with interquartile ranges) are shown in Figure 2 and the percentages of patients who attained the medical castration of testosterone ≤ 50 ng/dL are summarized in Table 4.
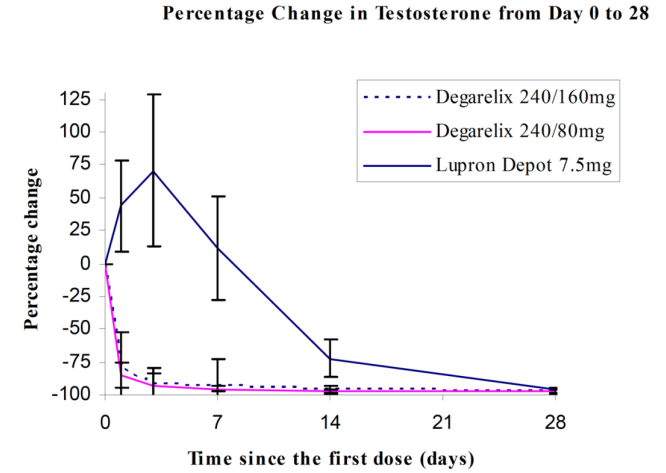
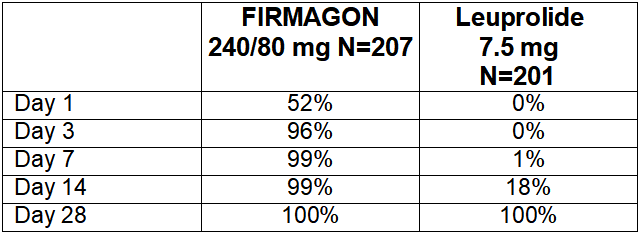
In the clinical trial, PSA levels were monitored as a secondary endpoint. PSA levels were lowered by 64% two weeks after administration of FIRMAGON, 85% after one month, 95% after three months, and remained suppressed throughout the one year of treatment. These PSA results should be interpreted with caution because of the heterogeneity of the patient population studied. No evidence has shown that the rapidity of PSA decline is related to a clinical benefit.
FIRMAGON is available as:
Store FIRMAGON at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). [see USP Controlled Room Temperature].
Advise the patient to read the FDA-approved patient labeling [see Patient Information].
Hypersensitivity
QT Interval Prolongation
Androgen Deprivation
Injection Site Reactions
Infertility
(degarelix for injection)
FIRMAGON is a prescription medicine used in the treatment of advanced prostate cancer. It is not known if FIRMAGON is safe or effective in children.
Do not receive FIRMAGON if you are allergic to degarelix or any ingredient in FIRMAGON. See the end of this leaflet for a complete list of ingredients in FIRMAGON.
Talk to your healthcare provider before receiving FIRMAGON if you have any of these conditions.
Before receiving FIRMAGON, tell your healthcare provider about all your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.
You will receive an injection of FIRMAGON from your healthcare provider.
FIRMAGON can cause serious side effects, including:
The common side effects of FIRMAGON include:
Other side effects include decreased sex drive and erectile function problems.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Medicines are sometimes prescribed for conditions that are not mentioned in a Patient Information leaflet. Do not use FIRMAGON for a condition for which it was not prescribed. Do not give FIRMAGON to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about FIRMAGON that is written for health professionals.
Active ingredient: degarelix (as acetate)
Inactive ingredient: mannitol
MANUFACTURED FOR:
FERRING PHARMACEUTICALS INC., PARSIPPANY, NJ 07054
Manufactured in Germany
For more information, go to www.FIRMAGON.com or call 1-888-337-7464.
This Patient Information has been approved by the U.S. Food and Drug Administration.
02/2020
MANUFACTURED FOR:
![]()
FERRING PHARMACEUTICALS INC.
PARSIPPANY, NJ 07054
Manufactured in Germany.
02/2020
8109000033


